HT 4
Morbus Canavan – eine Erbkrankheit
Morbus Canavan ist eine sehr seltene Erbkrankheit mit häufig schweren Symptomen. Trotz der Seltenheit ist die Krankheit gut untersucht. Dies führte zur Etablierung von Diagnoseverfahren und zur Entwicklung von Gentherapieansätzen, die zunächst in Tierversuchen getestet wurden.Leite den Erbgang von Morbus Canavan ab, indem du andere Erbgänge begründet ausschließt (M 1).
Gib alle möglichen Genotypen der Personen 1, 6 und 13 an (M 1).
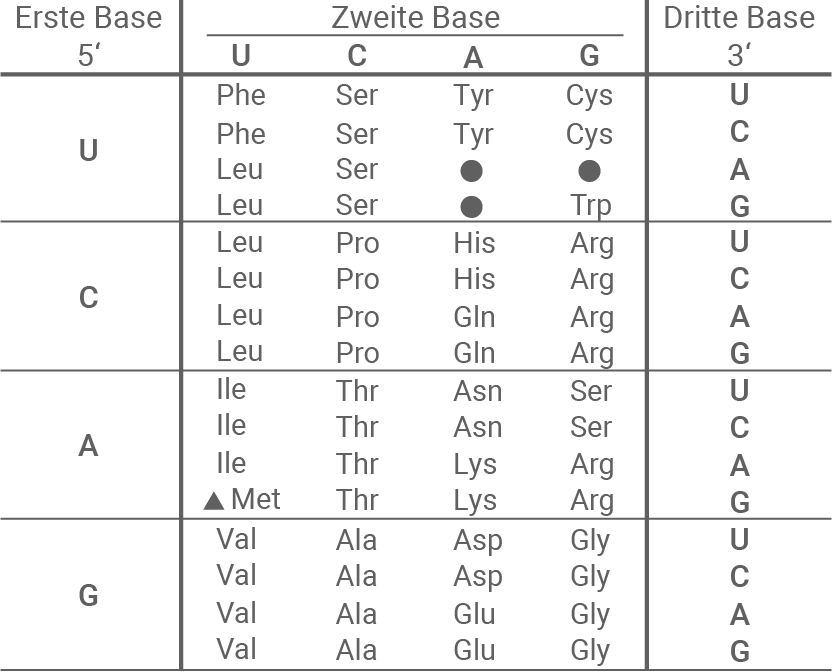
Leite für die in Tabelle 1 gezeigten DNA-Sequenzen die Aminosäuresequenzen sowie den Mutationstyp ab (M 2 und M 5).
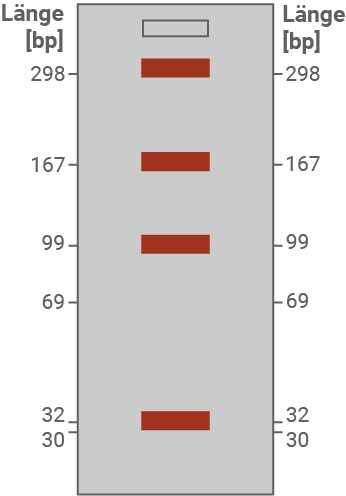
Erkläre das Grundprinzip einer DNA-Gelelektrophorese.
Erkläre die in Abbildung 2 dargestellten Ergebnisse (M 2 und M 3).
Skizziere in Abbildung 3 das zu erwartende Gelbild einer heterozygoten Person und begründe deine Skizze (M 1 bis M 3).
Fasse die in Abbildung 4 gezeigten Ergebnisse zusammen (M 4).
Beurteile den Erfolg der Gentherapie bei Ratten (M 1 bis M 4).
Diskutiere den Einsatz der in M 4 dargestellten Gentherapie bei Patienten mit Morbus Canavan (M 1 bis M 4).
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?M 1 Morbus Canavan
Morbus Canavan ist eine sehr seltene, genetisch bedingte Erkrankung. Betroffene Neugeborene weisen häufig keine Auffälligkeiten auf, zeigen jedoch in ihrer weiteren Entwicklung zum Teil schwere Symptome. Sie sind nicht in der Lage, Muskelspannung aufzubauen und eigenständige Bewegungen durchzuführen. Im Gehirn lässt sich ein zunehmender Abbau der Weißen Substanz feststellen. Als Weiße Substanz werden Teile des Zentralen Nervensystems bezeichnet, die aus Axonen bestehen. Die weiße Färbung entsteht durch die Myelinscheiden der Axone. Die meisten von Morbus Canavan betroffenen Kinder sterben noch vor dem Erreichen des 20. Lebensjahres an den Folgen der Erkrankung. Abbildung 1 zeigt einen Ausschnitt aus dem Stammbaum einer Familie, in der Morbus Canavan auftritt.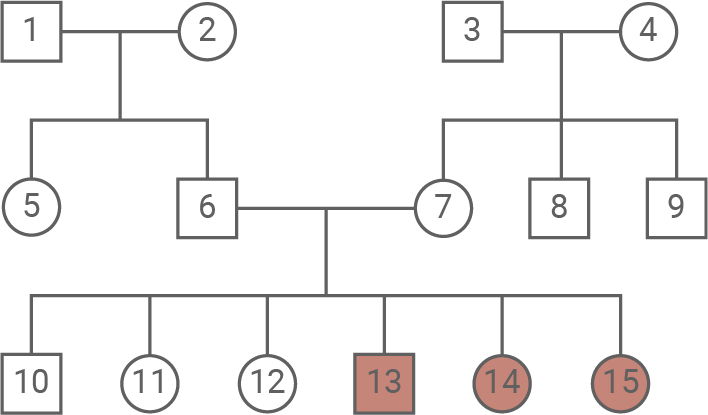
M 2 Genetische Ursachen
Mittlerweile sind bei Morbus Canavan über 50 verschiedene Mutationen im ASPA-Gen nachgewiesen worden. Das ASPA-Gen codiert für das Enzym Aspartoacylase (ASPA). Dieses Enzym katalysiert im Gehirn die Umsetzung von N-Acetyl-Aspartat (NAA) in Acetat und Aspartat. Acetat kann zu Acetyl-CoA umgesetzt werden. Acetyl-CoA spielt für die Myelinisierung der Axone eine wichtige Rolle.
M 3 Diagnose von Morbus Canavan
Bei einer Familie, in der das in Tabelle 1 dargestellte mutierte ASPA-Allel auftritt, wurde ein 298 Basenpaare langer Abschnitt des ASPA-Gens von betroffenen und nicht-betroffenen Personen durch PCR vervielfältigt. Die PCR-Produkte wurden anschließend mit dem Restriktionsenzym MboI geschnitten. Restriktionsenzyme schneiden spezifische Sequenzen doppelsträngiger DNA. MboI schneidet direkt vor der Sequenz 5' GATC 3'. Die PCR-Produkte wurden nach der Restriktion gelelektrophoretisch aufgetrennt und sichtbar gemacht (Abb. 2).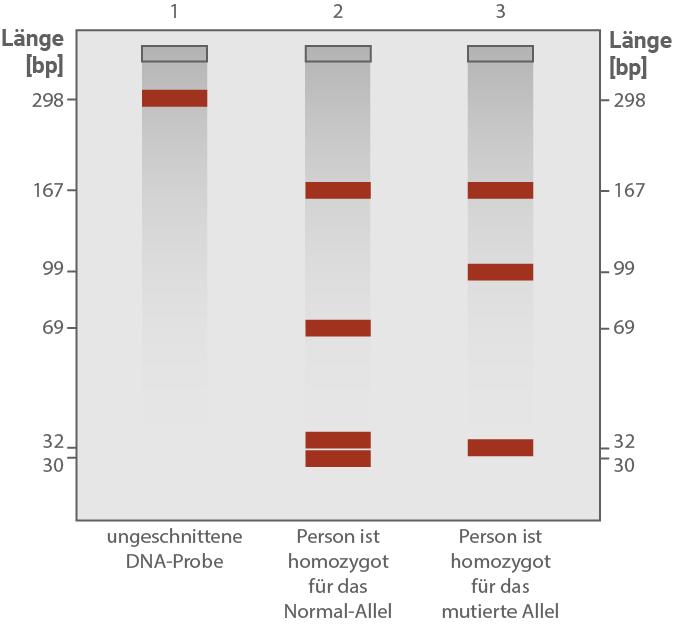
![Schematische vertikale Säule mit Skala Länge [bp] und Zahlen 298–30 sowie kleinem Rechteck oben](https://www.schullv.de/resources/images/mathe/desktop/11c121ed_vorlage-gelbild.png)
M 4 Gentherapie bei Ratten
Eine mögliche Gentherapie für Morbus Canavan wurde in Tierversuchen erprobt. Dazu wurden Ratten verwendet, denen das ASPA-Gen fehlte. Diesen ASPA-Ratten-Mutanten wurden genetisch veränderte Viren in das Gehirn injiziert, die nicht-mutierte Kopien des ASPA-Gens trugen. Dieses Vorgehen wird als Gentransfer bezeichnet. Die nicht-mutierten Genkopien wurden in die Zellkerne der Gehirnzellen eingeschleust und dort langfristig exprimiert. Der Einsatz solcher Viren kann in seltenen Fällen Abwehrreaktionen des Immunsystems hervorrufen. Anschließend wurden die Aspartoacylase-Enzymaktivität und die N-Acetyl-Aspartat-Konzentration (NAA-Konzentration) im Gehirn von Wildtyp-Ratten gemessen. Die ermittelten Werte wurden mit den jeweiligen Werten bei ASPA-Ratten-Mutanten ohne Gentransfer und bei ASPA-Ratten-Mutanten mit Gentransfer verglichen (Abb. 4). Die mit dem ASPA-Gentransfer behandelten ASPA-Ratten-Mutanten zeigten in motorischen Tests leicht verbesserte Balance- und Koordinationsfähigkeiten.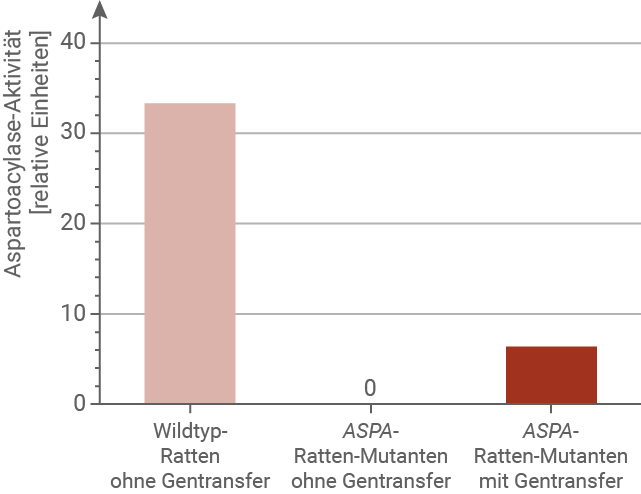
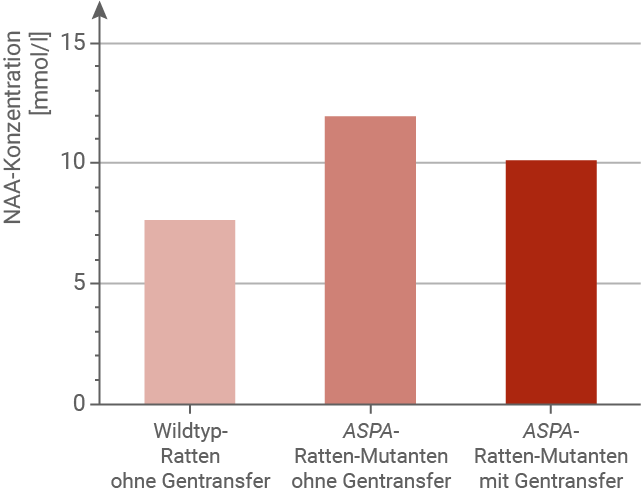
B die NAA-Konzentration
M 5 Codesonne und Tabelle zum genetischen Code
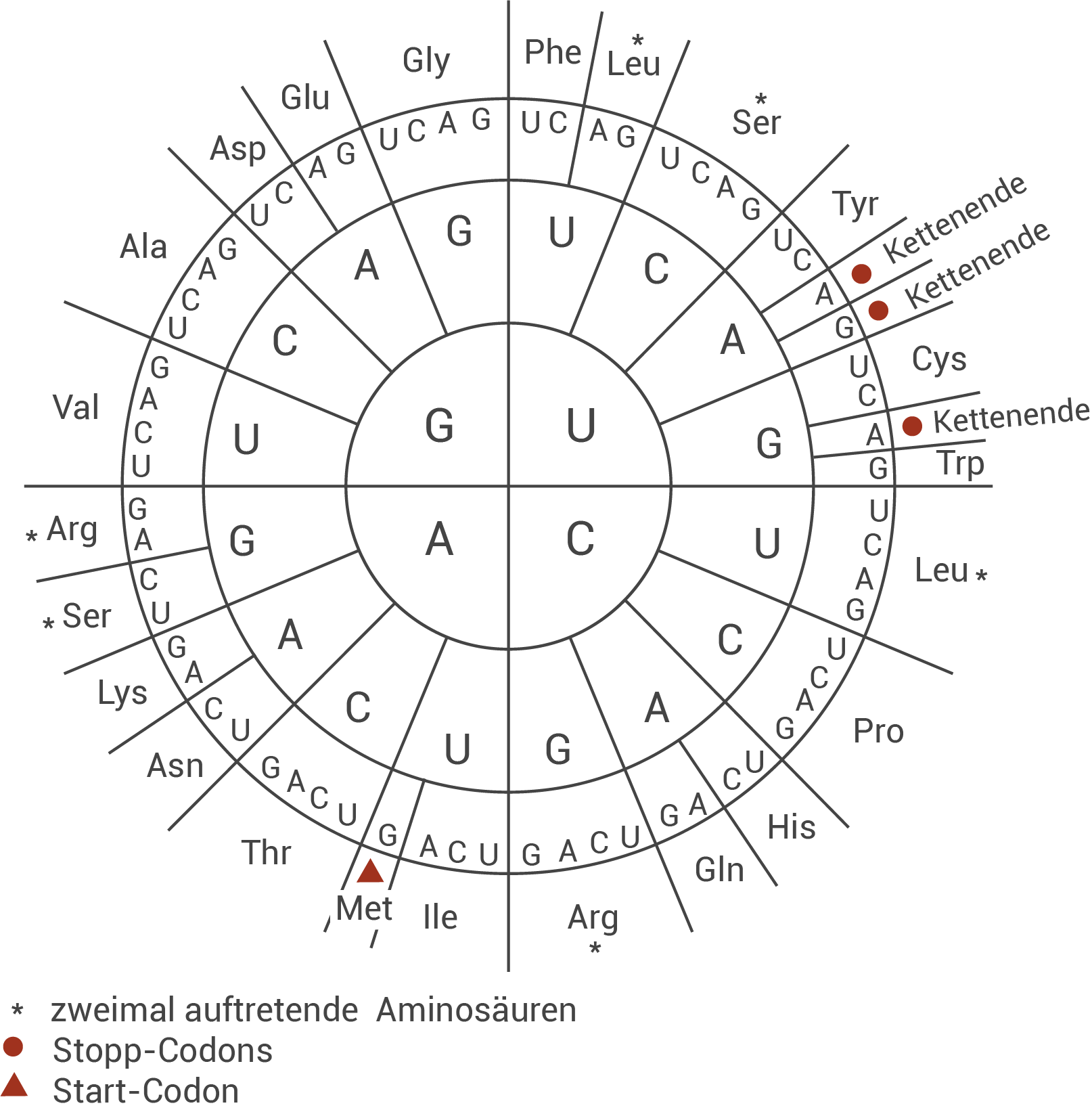
Arg Arginin
Asn Asparagin
Asp Asparaginsäure
Cys Cystein
Gln Glutamin
Glu Glutaminsäure
Gly Glycin
His Histidin
Ile Isoleucin
Leu Leucin
Lys Lysin
Met Methionin
Phe Phenylalanin
Pro Prolin
Ser Serin
Thr Threonin
Trp Tryptophan
Tyr Tyrosin
Val Valin
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?Analyse des Erbgangs:
Der Stammbaum zeigt, dass gesunde Eltern, etwa die Personen 6 und 7, ein betroffenes Kind (13) haben können. Das spricht für einen rezessiven Erbgang. Da sowohl Männer als auch Frauen betroffen sind, lässt sich eine X-chromosomale Vererbung ausschließen. Eine Y-chromosomale Vererbung ist unmöglich, weil auch Frauen betroffen sein können. Eine mitochondriale Vererbung scheidet aus, da betroffene Kinder nicht nur von betroffenen Müttern abstammen. Damit bleibt ein autosomal-rezessiver Erbgang. Person 13 ist betroffen und trägt daher den Genotyp aa. Person 6 ist phänotypisch gesund, hat aber ein betroffenes Kind; sie muss deshalb heterozygote Trägerin (Aa) sein. Person 1 ist gesund und kann genetisch AA oder Aa sein; beide Genotypen passen zum Stammbaum und lassen sich aus den gegebenen Informationen nicht eindeutig unterscheiden.Vergleich von Normal- und Mutantenallel:
Material ist der nichtcodogene DNA-Strang des Normal-Allels angegeben als CTG CAG GAT CAA GAC TGG. Die dazugehörige mRNA lautet: CUG CAG GAU CAA GAC UGG, woraus sich die Aminosäuresequenz Leu – Gln – Asp – Gln – Asp – Trp ergibt. Beim Mutantenallel ist nur das dritte Triplett verändert: GAT → GTT. Die mRNA enthält an dieser Stelle statt GAU jetzt GUU, sodass sich die Sequenz zu Leu – Gln – Val – Gln – Asp – Trp ändert. Damit liegt eine Punktmutation vor, genauer eine Missense-Mutation, weil ein Basenaustausch zur Einlagerung einer anderen Aminosäure (Asp → Val) führt.Zweck und Funktionsweise der DNA-Gelelektrophorese:
DNA-Gelelektrophorese dient dazu, DNA-Fragmente unterschiedlicher Länge aufzutrennen und sichtbar zu machen. DNA ist aufgrund der Phosphatgruppen im Zucker-Phosphat-Rückgrat insgesamt negativ geladen und wandert im elektrischen Feld in Richtung Anode. Das Agarosegel bildet ein poröses Netzwerk. Kleine Fragmente können sich leichter durch die Poren bewegen und wandern daher schneller und weiter als große Fragmente. So entstehen im Gel je nach Fragmentlänge unterschiedlich weit gewanderte Banden, die nach Färbung sichtbar gemacht und mit einem Größenstandard verglichen werden können. Auf diese Weise lassen sich Längenunterschiede nachweisen, zum Beispiel durch Mutationen oder unterschiedliche Restriktionsschnittmuster.Interpretation der Bandenmuster und heterozygoter Genotyp:
Das PCR-Produkt hat eine Länge von 298 bp.- In der ungeschnittenen Probe erscheint eine einzige Bande bei 298 bp.
- Beim homozygoten Normal-Allel schneidet MboI an zwei Erkennungsstellen im Fragment. Dadurch entstehen vier Fragmente unterschiedlicher Länge, die im Gel als vier Banden im unteren Bereich sichtbar sind (z. B. in der Nähe von ca. 167 bp, 99 bp, 69 bp und 32–30 bp; vgl. Abb. 2).
- Beim homozygoten Mutanten-Allel ist eine Restriktionsschnittstelle durch die Punktmutation verändert und wird nicht mehr erkannt. Dadurch entsteht ein anderes Fragmentmuster mit weniger Schnittstellen und entsprechend weniger Banden; im Material sind hier deutlich weniger Banden zu sehen als beim Normal-Allel.
Bewertung der Gentherapie anhand von Enzymaktivität und NAA-Konzentration:
In Abb. 4A weist der Wildtyp eine hohe Aspartoacylase-Aktivität (nahe 100 %) auf, während Knockout-Ratten ohne Therapie keine messbare Aktivität besitzen. Nach Gentherapie zeigen die behandelten Knockout-Ratten zwar wieder eine messbare Aktivität, diese liegt aber deutlich unter dem Wildtyp, ungefähr im Bereich von rund einem Fünftel bis einem Viertel der normalen Aktivität. In Abb. 4B ist die N-Acetyl-Aspartat-Konzentration (NAA) bei Knockout-Ratten stark erhöht. Nach Gentherapie sinkt der Wert zwar ab, bleibt aber klar über dem Niveau der Wildtyp-Tiere. Biochemisch gesehen verbessert die Gentherapie die Situation also etwas, stellt aber weder die Enzymaktivität noch die NAA-Konzentration vollständig wieder her. Zusammenfassend zeigt die Gentherapie nur eine begrenzte Wirksamkeit: Sie mildert die Stoffwechselstörung, erreicht aber keine annähernd vollständige Normalisierung.Chancen und Grenzen der Gentherapie bei Morbus Canavan:
Die Gentherapie greift kausal an der Ursache von Morbus Canavan an, indem sie das fehlende ASPA-Gen in Nervenzellen einbringt. Tierexperimente zeigen, dass damit die Enzymaktivität etwas steigt, die NAA-Konzentration sinkt und motorische Funktionen leicht verbessert werden. Gleichzeitig sind die Grenzen deutlich:- Die biochemischen Werte normalisieren sich nicht vollständig.
- Die motorischen Verbesserungen bleiben eher moderat.
- Es bestehen relevante Risiken wie Immunreaktionen gegen den viralen Vektor, eine unvollständige Verteilung im Gehirn und unklare Langzeitfolgen.
- Der Therapieerfolg hängt stark vom Zeitpunkt ab; bei spät behandelten Patienten sind bereits eingetretene Schäden durch fehlende Myelinisierung nur schwer oder gar nicht rückgängig zu machen.