HT 4
Morbus Canavan – eine Erbkrankheit
Morbus Canavan ist eine sehr seltene Erbkrankheit mit häufig schweren Symptomen. Trotz der Seltenheit ist die Krankheit gut untersucht. Dies führte zur Entwicklung von Gentherapieansätzen, die zunächst in Tierversuchen getestet wurden.Leite den Erbgang von Morbus Canavan ab, indem du andere Erbgänge begründet ausschließt (M 1).
Gib alle möglichen Genotypen der Personen 1, 6 und 13 an (M 1).
Stelle allgemein drei Genmutationen und jeweils eine mögliche Auswirkung auf das Polypeptid dar.
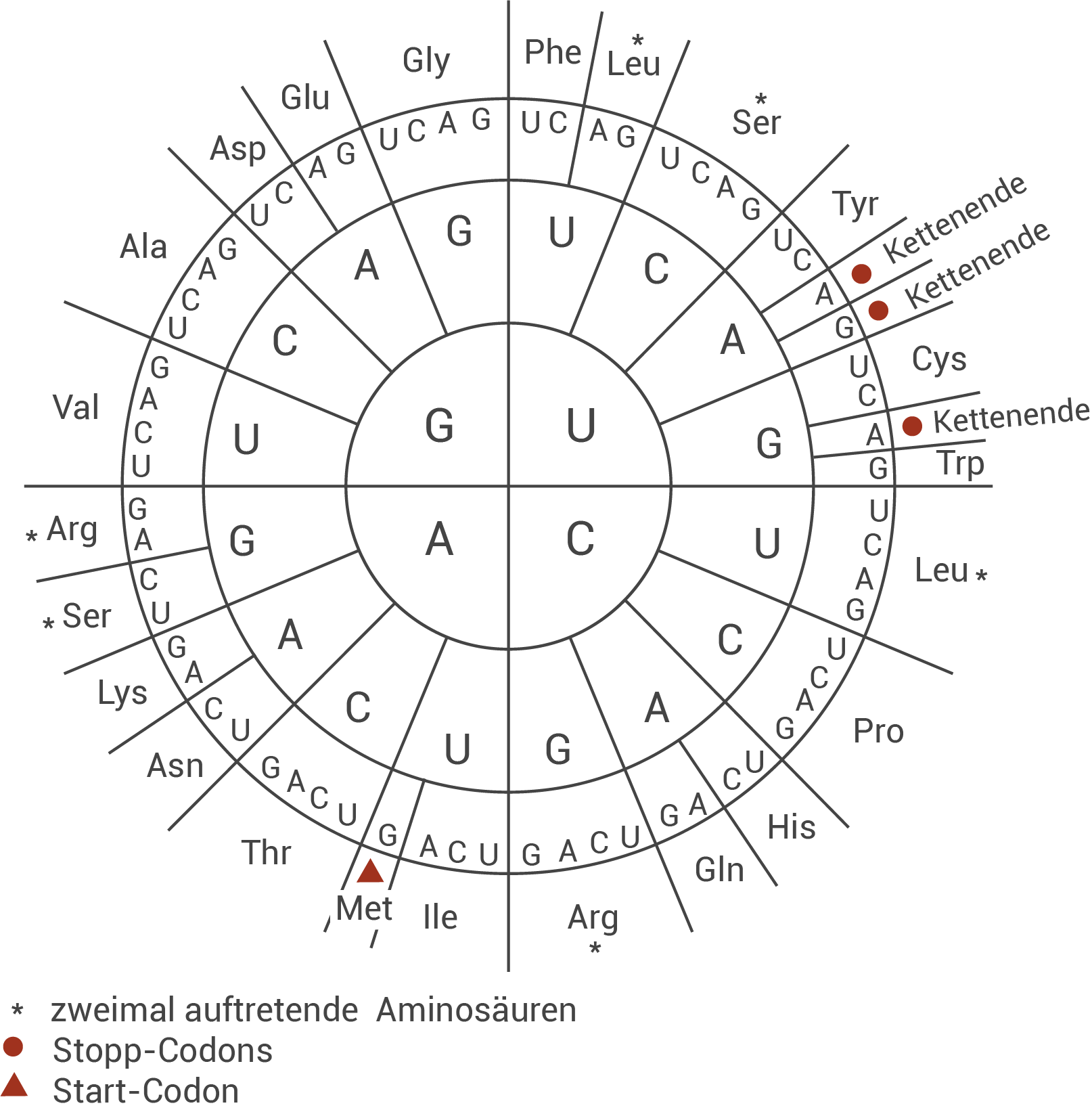
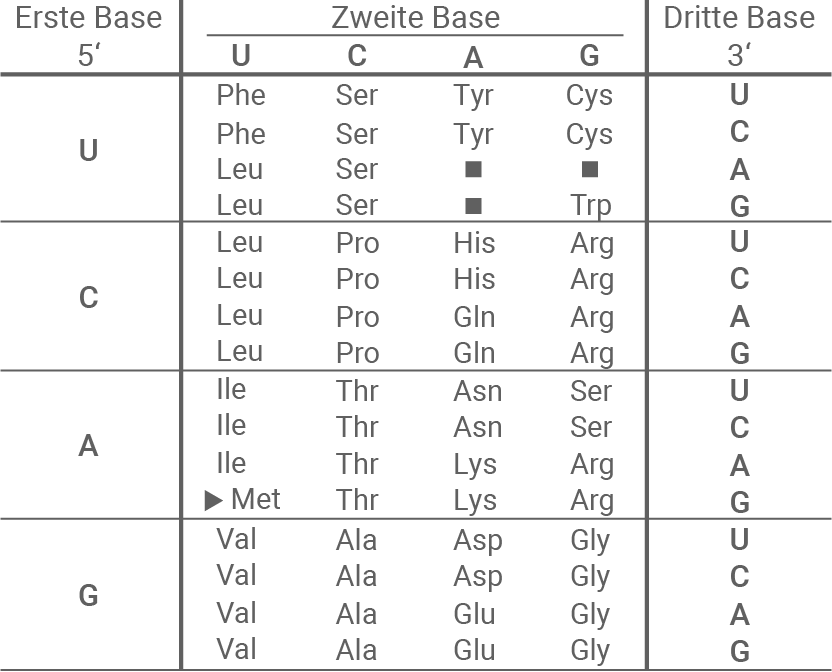
Leite für die in Tabelle 1 gezeigten DNA-Sequenzen die Aminosäuresequenzen sowie den Mutationstyp ab (M 2 und M 4).
Fasse die in Abbildung 2 gezeigten Ergebnisse zusammen (M 3).
Beurteile den Erfolg der Gentherapie bei Ratten (M 1 bis M 3).
Diskutiere den Einsatz der in M 3 dargestellten Gentherapie bei Patienten mit Morbus Canavan (M 1 bis M 3).
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?M 1 Morbus Canavan
Morbus Canavan ist eine sehr seltene, genetisch bedingte Erkrankung. Betroffene Neugeborene weisen häufig keine Auffälligkeiten auf, zeigen jedoch in ihrer weiteren Entwicklung zum Teil schwere Symptome. Sie sind nicht in der Lage, Muskelspannung aufzubauen oder eigenständige Bewegungen durchzuführen. Im Gehirn lässt sich ein zunehmender Abbau der Weißen Substanz feststellen. Als Weiße Substanz werden Teile des Zentralen Nervensystems bezeichnet, die aus Axonen bestehen. Die weiße Färbung entsteht durch die Myelinscheiden der Axone. Die meisten von Morbus Canavan betroffenen Kinder sterben noch vor dem Erreichen des 20. Lebensjahres an den Folgen der Erkrankung. Abbildung 1 zeigt einen Ausschnitt aus dem Stammbaum einer Familie, in der Morbus Canavan auftritt.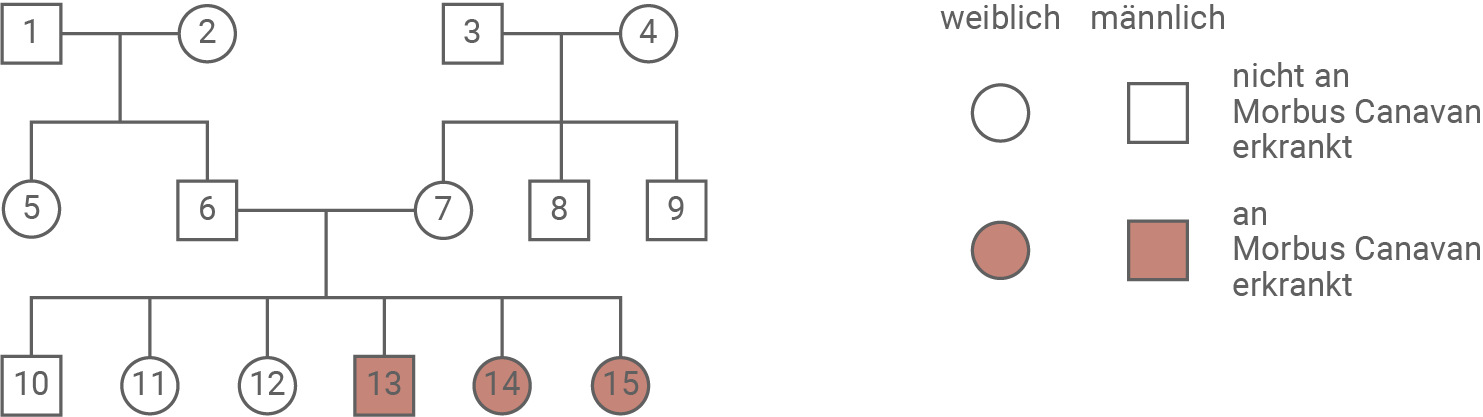
M 2 Genetische Ursachen
Mittlerweile sind bei Morbus Canavan über 50 verschiedene Mutationen im ASPA-Gen nachgewiesen worden. Das ASPA-Gen codiert für das Enzym Aspartoacylase (ASPA). Dieses Enzym katalysiert im Gehirn die Umsetzung von N-Acetyl-Aspartat (NAA) in Acetat und Aspartat. Acetat kann zu Acetyl-CoA umgesetzt werden. Acetyl-CoA spielt für die Myelinisierung der Axone eine wichtige Rolle. Tabelle 1 Ausschnitt aus dem nicht-codogenen DNA-Strang des ASPA-Gens einer nicht-betroffenen und einer betroffenen Person
M 3 Gentherapie bei Ratten
Eine mögliche Gentherapie für Morbus Canavan wurde in Tierversuchen erprobt. Dazu wurden Ratten verwendet, denen das ASPA-Gen fehlte. Diesen ASPA-Ratten-Mutanten wurden genetisch veränderte Viren in das Gehirn injiziert, die nicht-mutierte Kopien des ASPA-Gens trugen. Dieses Vorgehen wird als Gentransfer bezeichnet. Die nicht-mutierten Genkopien wurden in die Zellkerne der Gehirnzellen eingeschleust und dort langfristig exprimiert. Der Einsatz solcher Viren kann in seltenen Fällen Abwehrreaktionen des Immunsystems hervorrufen. Anschließend wurden die Aspartoacylase-Enzymaktivität und die N-Acetyl-Aspartat-Konzentration (NAA-Konzentration) im Gehirn von Wildtyp-Ratten gemessen. Die ermittelten Werte wurden mit den jeweiligen Werten bei ASPA-Ratten-Mutanten ohne Gentransfer und bei ASPA-Ratten-Mutanten mit Gentransfer verglichen (Abb. 2). Die mit dem ASPA-Gentransfer behandelten ASPA-Ratten-Mutanten zeigten in motorischen Tests leicht verbesserte Balance- und Koordinationsfähigkeiten.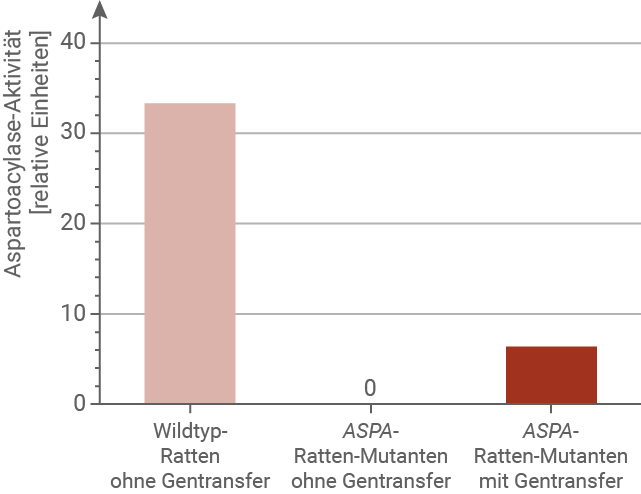
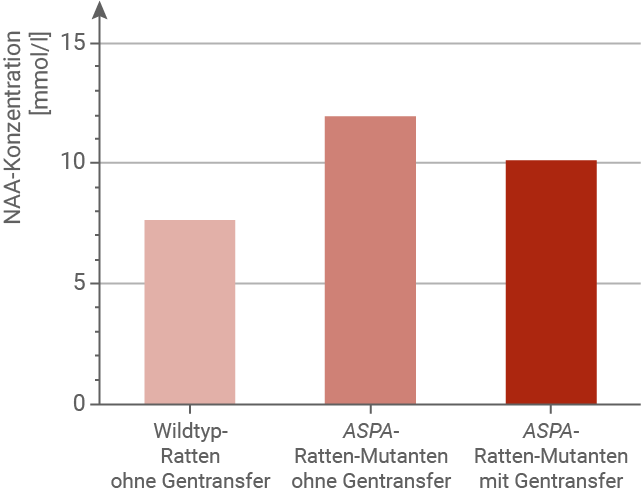
A die Enzymaktivität des Aspartoacylase-Enzyms und B die NAA-Konzentration
M 4 Codesonne und Tabelle zum genetischen Code
Arg Arginin
Asn Asparagin
Asp Asparaginsäure
Cys Cystein
Gln Glutamin
Glu Glutaminsäure
Gly Glycin
His Histidin
Ile Isoleucin
Leu Leucin
Lys Lysin
Met Methionin
Phe Phenylalanin
Pro Prolin
Ser Serin
Thr Threonin
Trp Tryptophan
Tyr Tyrosin
Val Valin
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?Morbus Canavan wird autosomal-rezessiv vererbt, da die Krankheit nur bei homozygot vorliegenden mutierten Allelen auftritt. Betroffene Kinder wie Person 13 müssen den Genotyp aa besitzen, da beide Allele defekt sind. Personen, die gesund sind, aber betroffene Kinder haben, wie Person 6, müssen heterozygot sein und somit den Genotyp Aa tragen. Bei Person 1 ist sowohl AA als auch Aa möglich, da das defekte Allel auch von Person 2 stammen könnte. Ein dominanter Erbgang ist auszuschließen, da gesunde Eltern sonst keine erkrankten Kinder haben könnten. Auch ein X-chromosomaler Erbgang ist ausgeschlossen, da sowohl Männer als auch Frauen betroffen sein können. Damit ist eindeutig, dass es sich um einen autosomal-rezessiven Erbgang handelt, bei dem 25 % der Kinder erkranken, wenn beide Eltern heterozygot sind. Person 13 trägt sicher aa, Person 6 sicher Aa, während Person 1 entweder AA oder Aa trägt. Andere nicht betroffene Geschwister können ebenfalls AA oder Aa sein. So erklärt sich das Auftreten der Erkrankung trotz meist gesunder Eltern.
Eine Missense-Mutation entsteht durch einen Basenaustausch, der zu einer veränderten Aminosäure im Protein führt. Dadurch kann die Struktur und Funktion des Proteins beeinträchtigt werden, z. B. durch eine veränderte Faltung. Eine Nonsense-Mutation entsteht ebenfalls durch einen Basenaustausch, jedoch entsteht hierbei ein vorzeitiges Stopcodon. Das Protein wird dadurch verkürzt und ist meist funktionslos. Eine Frameshift-Mutation entsteht durch Insertion oder Deletion von Basen, die nicht durch drei teilbar sind. Dadurch verschiebt sich das Leseraster und alle nachfolgenden Codons werden falsch abgelesen. Dies führt zu einer komplett veränderten Aminosäuresequenz ab der Mutationsstelle. Häufig entsteht dadurch ebenfalls ein verfrühtes Stopcodon, sodass das Protein stark verkürzt vorliegt. Missense-Mutationen haben oft eine abgeschwächte, aber noch teilweise vorhandene Enzymaktivität zur Folge. Nonsense- und Frameshift-Mutationen führen dagegen meist zu einem vollständigen Funktionsverlust des Proteins.
In Tabelle 1 sind die DNA-Sequenzen für das normale und das mutierte Allel dargestellt. Aus der DNA wird zunächst die komplementäre mRNA-Sequenz gebildet:
- normales Allel:
- DNA: …ATG GAA TCC…
- mRNA: …AUG GAA UCC…
- Aminosäuren: Met – Glu – Ser
- mutiertes Allel:
- DNA: …ATG GAA TGC…
- mRNA: …AUG GAA UGC…
- Aminosäuren: Met – Glu – Cys
Die in Abbildung 2 dargestellten Ergebnisse zeigen deutliche Unterschiede zwischen Wildtyp-Ratten, Mutanten und Mutanten nach Gentherapie. Bei Wildtyp-Ratten ist die Enzymaktivität der Aspartoacylase hoch und die NAA-Konzentration im Gehirn niedrig. Bei ASPA-Mutanten ist die Enzymaktivität deutlich reduziert und die NAA-Konzentration stark erhöht. Nach Gentherapie konnte die Enzymaktivität signifikant gesteigert werden, wenn auch nicht vollständig bis zum Wildtyp-Niveau. Parallel dazu sank die NAA-Konzentration im Gehirn der behandelten Tiere deutlich ab. Zusätzlich zeigten die behandelten Mutanten Verbesserungen in motorischen Tests, insbesondere in Balance und Koordination. Damit belegt die Gentherapie, dass die eingeschleusten Gene exprimiert und funktionsfähige Enzyme gebildet werden. Der Erfolg ist jedoch nicht vollständig, da die Enzymaktivität unter Wildtyp höher bleibt und nicht alle Symptome beseitigt werden. Dennoch ist die Therapie als insgesamt erfolgreich zu bewerten, da sie die Überlebenschancen und die Lebensqualität der Tiere verbessert. Langfristige Daten fehlen allerdings noch, sodass eine abschließende Beurteilung schwierig bleibt. Insgesamt zeigt sich eine deutliche Wirksamkeit der Gentherapie im Tiermodell, die Hoffnung für die Anwendung beim Menschen macht.
Die Gentherapie bei Morbus Canavan bietet eine vielversprechende Behandlungsoption, da bisher nur symptomatische Ansätze existieren. Durch den Ersatz des defekten ASPA-Gens kann das Enzym wieder produziert werden, wodurch die NAA-Akkumulation im Gehirn reduziert wird. Dies verbessert die Myelinisierung der Axone und lindert die neurologischen Symptome. Tierstudien haben gezeigt, dass die Enzymaktivität ansteigt, die NAA-Konzentration sinkt und motorische Fähigkeiten zunehmen. Ein Vorteil ist, dass die Therapie kausal ansetzt und nicht nur Symptome lindert. Gleichzeitig bestehen Herausforderungen wie die Überwindung der Blut-Hirn-Schranke, die Verteilung des Vektors im gesamten Gehirn und mögliche Immunreaktionen. Außerdem muss die Therapie frühzeitig erfolgen, da bereits zerstörtes Gewebe nicht regeneriert werden kann. Risiken wie unzureichende Transkription oder abnehmende Expression im Laufe der Zeit müssen ebenfalls berücksichtigt werden. Auch Sicherheitsaspekte wie Off-Target-Effekte durch den Vektor sind zu beachten. Insgesamt überwiegen jedoch die Chancen, da eine frühzeitige Gentherapie das Fortschreiten der Erkrankung deutlich verlangsamen könnte. Damit stellt sie eine der vielversprechendsten Strategien für diese seltene, schwer verlaufende Erbkrankheit dar.